
Significance
Cell wall assembly stalls when undecaprenyl pyrophosphate (UPP) cannot cycle back into the lipid carrier pool, and that interruption becomes especially consequential in resistant Gram-positive pathogens that already withstand many front-line drugs. The chemistry at issue here is not obscure: bacitracin binds UPP in a metal-dependent manner and blocks its dephosphorylation to undecaprenyl phosphate, a step bacteria need to keep peptidoglycan production moving. What makes the problem harder is not target validity but drug usability. Bacitracin has long had an unusual mechanistic position among peptide antibiotics because UPP is bacterial rather than mammalian, yet the parent natural product has not translated into broad systemic use due to a combination of factors already associated with bacitracin itself: modest antibacterial activity, a narrow Gram-positive spectrum, weak pharmacokinetic behavior, and resistance mediated through Bce-type ABC transporters that protect the target by disengaging bacitracin from UPP.
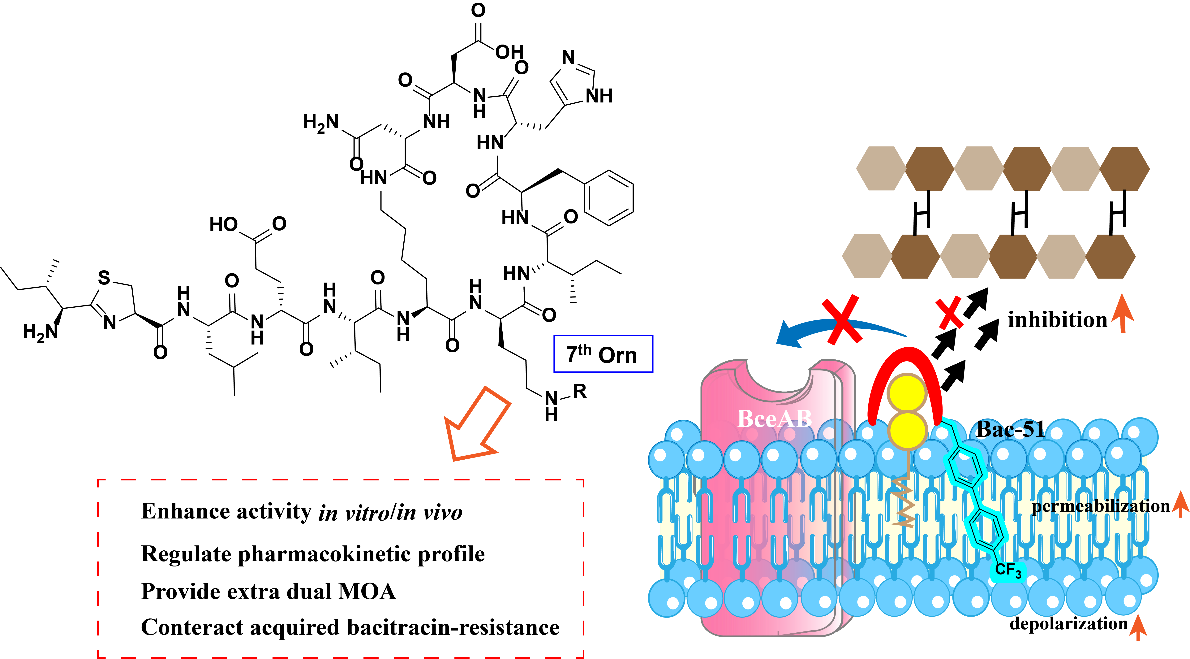
A joint research team, formed by Professor Dongliang Guan’s group from Yantai University, Shandong Laboratory of Yantai Drug Discovery, and Shanghai Institute of Materia Medica, Chinese Academy of Sciences, together with Professor Jinyong Zhang’s group from Army Medical University, has recently published their key research findings in Journal of Medicinal Chemistry, a top international journal in the field of medicinal chemistry. Sijie Cheng, Jingwen Liao served as the co-first authors of the paper. In this work, the researchers developed a regioselective reductive amination strategy that modifies the 7th ornithine δ-amino group of bacitracin and used it to prepare 53 new analogues with hydrophobic substituents. From that series, they identified Bac-51, a trifluoromethyl biphenyl lipopeptide analogue with improved activity against methicillin-, vancomycin-, daptomycin-, and bacitracin-resistant Gram-positive bacteria. Its distinct technical advance is that one defined modification site reshaped potency, pharmacokinetics, and mechanism at the same time. Mechanistically, the compound combines stronger interference with peptidoglycan precursor cycling and direct membrane-disrupting behavior, and it remains active against acquired bacitracin-resistant strains linked to BceAB overexpression.
The researchers began by establishing a regioselective reductive amination route that modified the δ-amino group of the 7th ornithine residue directly from commercial bacitracin, and they verified site selectivity by NMR signal shifts assigned to the ornithine side chain. That synthetic control mattered because the whole premise depended on changing one exposed amino group without disturbing the metal-binding and pyrophosphate-recognition features at the N-terminus. Under optimized conditions, the method generated 53 derivatives carrying aliphatic, aryl, and biaryl substituents, which gave the authors a sufficiently broad set to read local structure–activity relationships rather than relying on a single lead from the start. Screening against methicillin- and vancomycin-resistant S. aureus and several vancomycin-resistant Enterococcus strains showed a consistent gain in potency after hydrophobic installation at 7th ornithine. The pattern was chemically informative: alkyl substituents produced a chain-length dependence, aromatic substitution geometry affected activity, and biphenyl-containing analogues were especially strong. Bac-51, which carries a trifluoromethyl-substituted biphenyl group, emerged as the best compound, with activity improvements reported as 8- to 256-fold over bacitracin and measurable advantages over vancomycin and daptomycin in the resistant Gram-positive panel. That distribution of results gives the 7th ornithine position a mechanistic meaning beyond simple derivatization convenience; the site can transmit changes in appended hydrophobic character into large changes in antibacterial output. Time-kill studies added another layer. Bac-51 produced slow, concentration-dependent killing against MRSA USA300 LAC and stronger growth suppression against VRE E. faecium than bacitracin at matched testing logic, and testing against 30 clinical resistant isolates across seven Gram-positive species kept the same direction of activity.
The authors then followed the lead compound through safety, exposure, efficacy, and mechanism. In HEK-293 and HepG2 cells, Bac-51 showed no significant cytotoxicity at the tested concentrations; mouse erythrocytes showed no obvious hemolysis at 64 μg/mL; and mice tolerated intravenous doses of 50 and 100 mg/kg. Pharmacokinetic work in BALB/c mice after a single 10 mg/kg intravenous dose showed slower clearance than bacitracin, an approximately 1.8-fold increase in AUC, much higher plasma concentration at 4 h, and a larger steady-state distribution volume. That part of the study is conceptually important because it ties the same 7th ornithine modification site not only to potency but also to drug exposure, which is a different property altogether. In the lethal MRSA sepsis model, a single intravenous dose produced 30% survival at 20 mg/kg and 70% survival at 40 mg/kg, compared with 0% and 10% for bacitracin. Mechanistic experiments then explained why the lead behaved differently. Bac-51 drove greater accumulation of Park’s nucleotide than bacitracin, about 1.6-fold in the MRSA252 assay, which is consistent with stronger interruption of cell wall precursor flow. At the same time, membrane assays showed marked permeabilization and depolarization at concentrations far below those needed for bacitracin. Cardiolipin antagonized Bac-51 activity, whereas phosphatidylglycerol and phosphatidylethanolamine did not, and molecular modeling placed the trifluoromethyl biphenyl appendage toward the lipid-facing region of the complex. Electron microscopy then captured extensive damage to S. aureus cellular morphology after treatment. The design choice made at 7th ornithine did more than decorate the scaffold: it preserved UPP-directed chemistry and added membrane engagement, so the lead moved as a dual-acting lipopeptide rather than a refined copy of bacitracin. Serial passage completed the picture. Across 20 passages, Bac-51 held its MIC at 4 μg/mL, remained active against acquired bacitracin-resistant strains from S. aureus and E. faecalis, and qPCR linked the comparator resistance phenotype to BceAB overexpression.
What gives the new study weight is not only that Bac-51 is active. The stronger point is that the work of Professor Dongliang Guan and colleagues redefines where bacitracin can be productively engineered. For a long time, the molecule’s value has been tied mainly to its pyrophosphate-binding mechanism, with structural attention concentrated elsewhere on the scaffold. Here, the 7th ornithine side chain becomes a chemically intelligible control point for several properties at once: antibacterial potency, pharmacokinetic behavior, in vivo efficacy, and mechanism expansion. That is a meaningful shift in design logic. It says that semisynthetic improvement of a classic peptide antibiotic need not proceed by protecting the original mechanism in a rigid way; it can proceed by adding a second physical interaction mode, provided that the original recognition event remains accessible.
The mechanistic interpretation also matters beyond this particular analogue. Bac-51 did not simply bind harder to the old target in a generic sense. The data support a more specific picture in which appended hydrophobic structure changes how the compound occupies the interface between lipid-linked cell wall precursors and the bacterial membrane. Park’s nucleotide accumulation places the compound squarely in the cell-wall pathway, whereas permeabilization, depolarization, cardiolipin dependence, and modeling orient the added biphenyl unit toward membrane participation. That combination gives the field a practical example of how an established cell-wall antibiotic scaffold can be reworked into a dual-function agent without abandoning its original biochemical entry point. In resistant Gram-positive infections, that kind of built-in mechanistic breadth is scientifically meaningful because resistance systems that handle one interaction mode may not process the full composite state in the same way. The BceAB-related results fit that logic closely.
There is also a broader methodological message running through the paper. The authors did not treat structure–activity relationships as a side table appended to lead discovery. Yantai University, Shandong Laboratory of Yantai Drug Discovery, Shanghai Institute of Materia Medica and Army Medical University researchers used SAR to expose an overlooked residue as a site that regulates multiple downstream behaviors. That makes the study relevant to medicinal chemistry well beyond bacitracin itself. It gives a worked example of how a single, carefully chosen semisynthetic handle can connect molecular recognition, membrane physics, exposure, and resistance behavior inside one optimization campaign. The downstream implication, kept within the paper’s demonstrated scope, is that bacitracin-derived lipopeptides can be developed as systemic candidates against multidrug-resistant Gram-positive pathogens in a way that earlier bacitracin chemistry had not established. Bac-51 is the clearest expression of that idea in this study: a 7th-ornithine-modified analogue that couples retained UPP targeting with added membrane activity and sustained performance against bacitracin-resistant phenotypes.

Reference
Cheng S, Liao J, Chen Z, Li F, Zhu Y, Zhang J, Guan D. Novel Rationally Designed Lipopeptides Derived from Bacitracin: Combating Multidrug Resistance and Evading Bacitracin Resistance via Potentiated Cell Wall and Membrane Inhibitions. J Med Chem. 2025;68(15):16410-16426. doi: 10.1021/acs.jmedchem.5c01285.
Go to Journal of Medicinal Chemistry