
Significance
Cancer immunotherapy changed the treatment landscape by making durable tumor control possible through activation of the host immune system rather than direct cytotoxic attack alone. Among the major advances in oncology, immune checkpoint blockade is particularly attractive because antibodies targeting inhibitory pathways such as PD-1, PD-L1, and CTLA-4 showed that the “immune brake” of antitumor immunity could be released in a clinically meaningful way in at least a subset of patients. They also made clear that the immune system is not a secondary participant in cancer therapy, but in many settings a decisive determinant of whether tumor control can be sustained, extended to disseminated disease, or lost altogether. Checkpoint-based immunotherapy produces excellent clinical benefit in some patients but much weaker effects (70%~ 80%) in many others, and that uneven performance has remained one of the problems/grand challenges in the field. Several theories tried to explain this including T-cell exhaustion, insufficient neoantigen availability, and strongly immunosuppressive tumor microenvironments. Each of those factors carries weight, however, none fully resolves the basic question of why releasing antitumor inhibitory signaling can generate deep clinical responses in some tumors while producing only modest benefit in others. The issue becomes especially important in solid tumors, where immune priming, antigen presentation, and local suppression rarely evolve in a coordinated way.
A recurring difficulty is that successful immunotherapy requires more than the presence of T cells in or around a tumor. It depends on the presence of T cells that have actually acquired the capacity to recognize tumor-associated features and respond productively after stimulation. When that population is limited, the therapeutic value of checkpoint release or immune co-stimulation is limited from the outset. The authors[1] argue that many antigen-specific T cells remain functionally ineffective because they have not undergone adequate education through dendritic-cell presentation of tumor-derived material. A more effective strategy may require generating a larger supply of immunogenic tumor debris first, so that new tumor-recognizing cytolytic T cells can be primed before immunotherapy is expected to exert its full effect.
That reasoning creates a strong case for combining immunotherapy with a local treatment capable of producing both tumor destruction and immune-relevant neoantigen release. Phototherapy is attractive in that regard because it can damage cancer cells directly while also altering the inflammatory and antigenic environment within the tumor. In a recent research paper published in ACS Nano,[1] Dr. Munusamy Shanmugam, Dr. Chi-Shiun Chiang, and Professor Kuo Chu Hwang from the National Tsing Hua University, Taiwan, developed PEG-folate-functionalized LaB6 nanoparticles that absorb across NIR-I through NIR-IV and support wavelength-resolved phototherapy within one materials platform. They paired those nanoparticles with 2240 nm irradiation and anti-OX40 to create a local-to-systemic treatment strategy that couples tumor photodynamic destruction to immune co-stimulation. What is new here is the use of NIR-IV photodynamic priming to generate in situ whole-tumor vaccine material and convert a weak anti-OX40 response into strong control of remote and metastatic melanoma. They also framed the mechanism with a “blind T cells” model (see Scheme 1) that links poor immunotherapy performance to an upstream shortage of newly primed tumor-recognizing cytolytic T cells.
The research team first built the therapeutic system around LaB6 nanoparticles modified with PEG-folate, and the choice carried two immediate experimental consequences: prolonged circulation after intravenous dosing and stronger uptake by folate-receptor-expressing melanoma cells. The investigators measured a blood half-life of about 20 hours for the PEG-folate particles, compared with 9 hours for unmodified LaB6, and they detected higher tumor accumulation at 24 hours, which is why they fixed that time point for irradiation. That scheduling detail mattered because earlier irradiation would have obscured the effect of tumor enrichment of the nanomaterial. The authors then used the same nanoparticle platform to compare 808, 1064, 1550, and 2240 nm NIR light photo-excitation. They observed singlet oxygen generation at 1064 nm, whereas 1550 and 2240 nm produced strong hydroxyl-radical signals, even under hypoxic conditions, far above the values measured at 1064 and 808 nm. This distinction is relevant because hypoxia often weakens classical photodynamic strategies. Here, the longer-wavelength channels did not depend on the same oxygen chemistry, and that gave the NIR-III and NIR-IV conditions a different biological footing from the start. The study examined cell killing under those irradiation modes and found that ROS-producing conditions depressed melanoma viability more strongly than 808 nm treatment, which behaved mainly as photothermal heating. The comparison also brought out an instructive trade-off: 808 and 1550 nm produced larger temperature rises, but the deepest antitumor gains did not track peak heating alone.
The investigators carried that logic into immunogenic cell death. They observed that photodynamic conditions drove tumor death mainly through necrosis-dominant patterns, while 808 nm treatment yielded a more mixed necrotic-apoptotic profile. They then measured CRT exposure, HMGB1 release, and ATP release and found all three increased after photoirradiation, with the 2240 nm condition producing the highest DAMP levels. The research group also measured light transmission through tissue and recorded transmission reached 46% at 2240 nm, compared with 26% at 808 nm. That tissue-penetration gradient gives a plausible physical reason why the longest wavelength generated the strongest immunogenic output in the tumor model: more usable light survived tissue passage, so more nanoparticles could participate in chemistry deeper in the lesion.
The mouse studies tied these physical and cellular results to antitumor behavior. The researchers implanted primary and remote B16BL6 tumors and irradiated only the primary site. Without anti-OX40, all nanoparticle-plus-light groups slowed primary growth, but remote control stayed modest. After adding anti-OX40, remote suppression improved sharply, with the strongest effect at 2240 nm; median half-lifespan extended from 21 days to 83 days. The study also examined lung metastasis qualitatively and found visible metastatic nodules essentially absent in the 1550 and 2240 nm combination groups. When the investigators repeated key experiments in immune-deficient SCID mice, the therapeutic benefit dropped to roughly half of what they observed in immunocompetent animals. That loss is telling, because it argues that direct tumor phototoxicity was not carrying the whole treatment effect. The boosted outcome depended heavily on intact adaptive immunity.
The authors measured immune profiling of serum TNF-α, IFN-γ, IL-2, and IL-12 and found strong increases in the nanoparticle-plus-light-plus-anti-OX40 groups, especially under 1550 and 2240 nm irradiation. They also recorded stronger dendritic-cell maturation and larger populations and enhanced IFN-g production activity of CD8+ T cells, while the paper’s discussion attributes the combined therapy to simultaneous expansion of antitumor compartments and suppression of Treg and M2-type macrophage activity. Anti-OX40 alone could move the system somewhat, but once the photodynamic treatment generated a much larger supply of immunogenic tumor neoantigens, the agonist operated in a very different immune setting. Professor Kuo Chu Hwang and colleagues demonstrated that many less effective immunotherapy responses may be interpreted too narrowly when viewed mainly through checkpoint biology. Their data push attention upstream, toward antigen availability, dendritic-cell maturation, inflammatory cytokines, and the generation of freshly primed tumor-recognizing cytolytic T cells first, instead of removing the “immune brake” of the existing, but “blind” CD8+ T cells.
Long-wavelength photomedicine often draws attention because of penetration depth, but the new work links penetration to a specific immunological consequence: deeper light delivery produces more extensive, more immunogenic tumor destruction, which then changes how well an agonist immunotherapy can work at distant, untreated sites. The authors went beyond showing that 2240 nm light can damage a tumor and built a case that wavelength choice can alter the quality of the immune conversion triggered by local treatment. If that principle survives in larger and more heterogeneous tumor models, it could influence how photodynamic systems are designed when systemic immune cooperation is the real therapeutic goal. The study gives timing and sequence a more mechanistic role than they often receive in combination-therapy design. Anti-OX40 was not simply added to phototherapy; it was placed into a setting where tumor killing had already generated antigenic debris and inflammatory signals. That order is important because co-stimulation without sufficient antigen priming can only do so much. One can imagine this logic informing combinations with checkpoint inhibitors, cytokine agonists, radiation, chemotherapy, or other cell-killing modalities, though any such extension would need careful testing because the balance among antigen release, suppressive myeloid cells, and T-cell priming is unlikely to remain constant across tumor types.
By using one nanoparticle family across four near-infrared windows, the authors reduced a common source of ambiguity in comparative phototherapy papers. The wavelength-dependent differences can be read with more confidence because the photosensitizing platform stayed materially consistent. That makes the ranking of 2240 nm above 1550 nm, and both above 1064 and 808 nm in this melanoma system, more persuasive than a comparison built from unrelated agents. Whether the same ordering will persist in thicker tumors, other anatomical settings, or clinically practical light-delivery conditions remains to be determined, but the central point is clear: immunotherapy becomes more effective when local tumor killing first expands the pool of tumor-recognizing T cells.
Overall, the “blind T cells” concept can be illustrated in the Scheme I. Large majority of existing CD8+ T cells in cancer patients are actually “blind”, meaning cannot recognize tumor cells. Under such a condition, removing the “immune brake” by using immunotherapy agents will not improve the cancer cell killing efficacy, but leads to various autoimmune adverse effects. The “blind T cells” model suggests shifting the paradigm of cancer treatments to creation of new tumor-recognizing cytolytic T cells before removal of the “immune brake” using immunotherapy agents. Clinically, medical doctors in Taipei Veterans General Hospital in Taiwan observed that among 107 patients with advanced EGFR-mutant non-small cell lung cancer (NSCLC), the mediam number of patients treated with combined chemotherapy and immunotherapy has a longer overall survival of 20 months than 16 months from those treated with immunotherapy alone.[2] Medical doctors in Chang Gung Memorial Hospital at Linkou, Taiwan, also observed that among 137 human patients with liver cancer at late stage B or C hepatocellular carcinoma, the two year overall survival rate for patients treated with combined proton radiotherapy and immunotherapy is 77%, which is significantly higher than 42.7% observed from patients treated with proton-Tyrosine Kinase Inhibitor group and 52.7% from patients treated with proton radiotherapy alone.[3] These observations in clinical human cancer patients are supportive data for the validity of the “blind T cells” model. The “blind T cells” model proposes a new strategy to overcome the grand challenge of ineffectiveness of immunotherapies by shifting the paradigm of cancer treatments to generation of a massive number of newly created, tumor-recognizing cytotoxic T cells before immunotherapy is administered, instead of simple release of “immune brake” by immunotherapy alone.
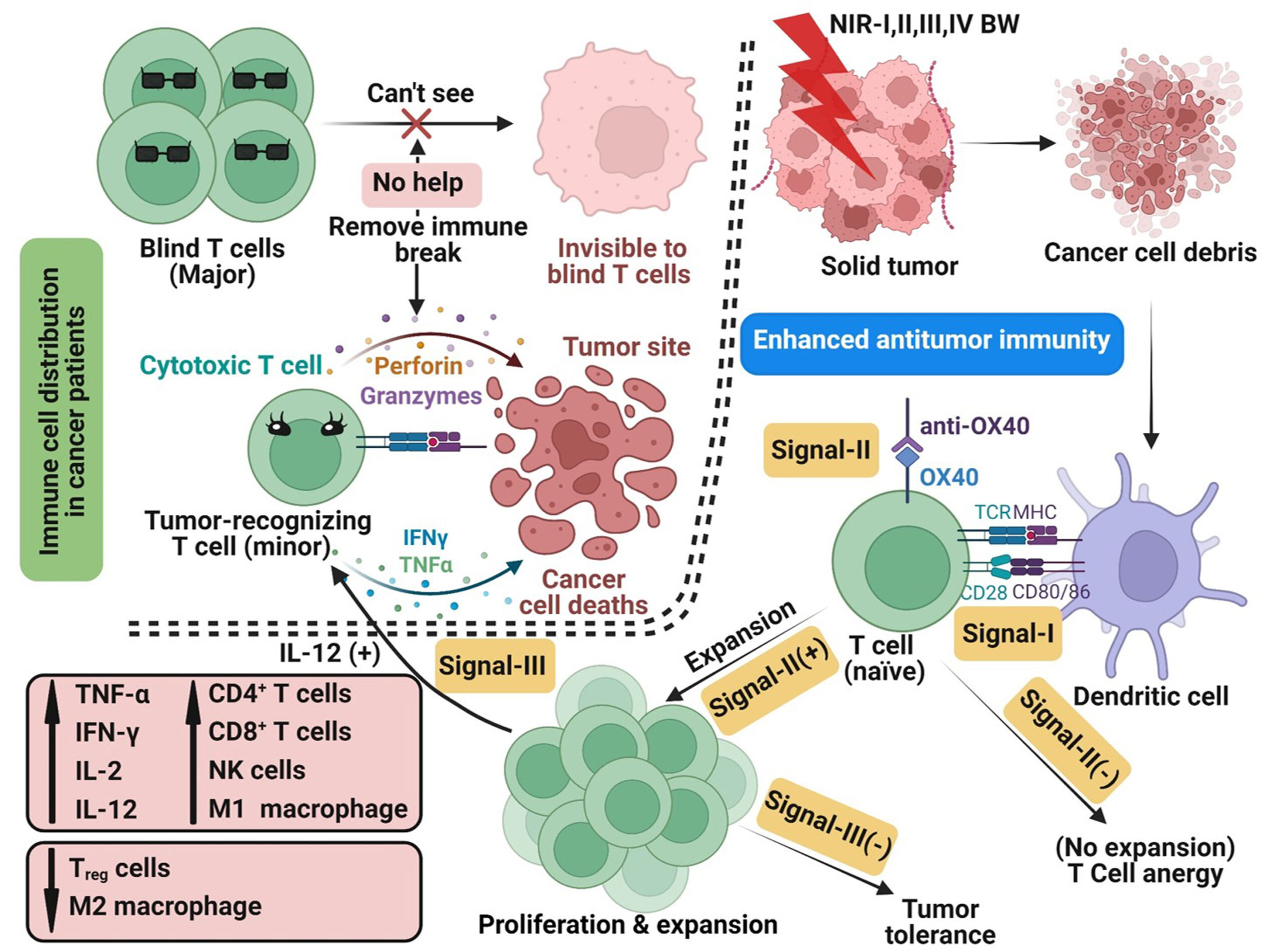
Scheme 1. A “Blind T Cells” Model to Rationalize the Ineffectiveness of Immunotherapies, and a Strategy to Reverse Ineffectiveness of Immunotherapies via Combination of the In Situ Generated Whole Cancer Cell Vaccine by NIR-I/-II/-III/-IV PDT with Immunomodulator Anti-OX40. [Image credit: ACS Nano. 2025;19(41):36129-36147. doi: 10.1021/acsnano.5c04323].
