
Significance
Loss of endoplasmic reticulum membrane area emerges early in adulthood, with cisternal sheets thinning, fragmenting, and giving way to sparse tubular networks as cells age. Such behavior poses a basic problem for cell biology because of the importance of endoplasmic reticulum in supporting protein translocation, folding, lipid synthesis, calcium handling, and signaling coordination, all processes that demand surface area, luminal volume, and spatial organization. A sustained reduction in ER mass appears incompatible with long-term homeostasis, however, ageing cells repeatedly tolerate and even reproduce this pattern. Most discussion of ER function during ageing has concentrated on signaling pathways that respond to protein misfolding. That focus has clarified how cells detect stress and adjust transcriptional programs, but it has left the material state of the organelle itself largely unexamined. ER morphology varies widely across cell types, reflecting functional demand. Rough sheets accommodate ribosome loading and protein maturation, whereas curved tubules favor lipid handling and membrane exchange. Changes in the balance between these architectures carry direct consequences for cellular output. Despite this, ageing has usually been treated as a backdrop against which ER stress responses act, not as a condition that reshapes ER structure itself. Several observations hint that this framing may be incomplete. Protein synthesis declines during normal ageing in many organisms, yet similar reductions accompany interventions that extend lifespan. Lipid metabolism often persists or increases under those same conditions. These trends suggest coordinated reallocation of ER capacity rather than passive decay. Structural remodeling could provide a means to execute such reallocation, though direct evidence across tissues and species has remained limited. One reason lies in technical constraints. Overexpressed ER markers distort subdomain identity, and static imaging misses gradual transitions. Selective autophagy of the ER offers a plausible mechanism for controlled remodeling. ER-phagy targets defined membrane regions for degradation, using receptors embedded in the ER itself. Studies in yeast and cultured cells have catalogued components of this machinery, often under nutrient limitation or pharmacological inhibition of growth signaling. Those same conditions repeatedly correlate with extended lifespan. Whether ER-phagy participates in normal ageing, and whether it contributes actively to physiological adjustment rather than damage control, has not been clear.
A recent research paper published in Nature Cell Biology and conducted by Dr. Eric Donahue, Dr. Nathaniel Hepowit, Dr. Elizabeth Ruark, Dr. Alexandra Mulligan, Dr. Brennen Keuchel, Dr. Nicholas Urban, Dr. Li Peng, Dr. Stedman Stephens, Dr. Derek Johnson, Dr. Natalie Wallace, Lauren Jackson, Professor Mark Ellisman, Rafael Arrojo e Drigo, Assistant Professor Andrew Folkmann, Dr. Matthias Truttmann, Dr. Jason MacGurn, and led by Professor Kristopher Burkewitz from the Department of Cell and Developmental Biology at Vanderbilt University School of Medicine, the authors developed endogenous, subdomain-specific ER reporters suitable for long-term in vivo imaging. They established quantitative metrics linking ER geometry, volume, and protein composition during ageing. The study defined ER-phagy as a driver of structural remodeling rather than a byproduct of stress. It identified TMEM-131 as a previously unrecognized regulator coordinating tissue-specific ER turnover.
Briefly, the research team established endogenous fluorescent markers to distinguish rough and tubular ER without overexpression artifacts. They generated genomic fusions to SEC-61.B to label ribosome-rich sheets and to the reticulon RET-1 to track curved tubules and sheet edges. Super-resolution imaging confirmed segregation of these markers in vivo, with patterns matching known ER specialization across tissues. And using these reporters, the investigators monitored ER architecture during adulthood in Caenorhabditis elegans. In young animals, hypodermal cells displayed dense cisternal networks with limited tubular connectivity. As animals aged, the same cells showed pronounced loss of ER signal, fragmentation of sheets, and expansion of reticular structures. Quantitative analysis revealed large reductions in ER footprint and fluorescence intensity alongside increased perimeter-to-area ratios, reflecting both reduced volume and altered geometry. The authors verified that these trends did not arise from imaging artifacts or global protein loss. Immunoblotting of epitope-tagged ER proteins reproduced the decline in ER mass. Mitochondrial markers remained stable, constraining the effect to the ER. Daily imaging demonstrated that much of the change occurred within the first days of adulthood, well before late-life failure. To relate structure to function, the study examined age-dependent proteomic datasets. ER-resident proteins involved in protein translocation, folding, and quality control declined broadly with age, paralleling loss of rough ER. In contrast, many lipid-associated ER proteins persisted or increased. This redistribution aligned with the observed morphological shift, linking geometry to metabolic emphasis rather than treating the two independently.
The researchers extended analysis across tissues. Intestinal cells, muscle, neurons, and hypodermis all exhibited ER reduction and remodeling, though the magnitude and subdomain composition varied. RET-1 levels nearly vanished in aged intestine, whereas other tubulating factors remained, implying compensation rather than uniform collapse. Male animals followed similar trajectories, excluding reproductive burden as a cause. Plus, electron microscopy reinforced these conclusions at ultrastructural resolution. Stacked rough sheets in young cells gave way to sparse, curved membranes in aged counterparts. Comparable patterns appeared in yeast during chronological ageing and in mammalian tissues, including mouse brain, liver, and skin, where ER protein abundance and footprint declined with age. To address mechanism, the study examined ER-phagy components. Genetic disruption of core autophagy machinery blocked age-associated ER remodeling. The authors identified TMEM-131 as a tissue-specific regulator required for ER turnover and connected ER-phagy activation to the IRE-1–XBP-1 branch of the unfolded protein response. Longevity paradigms that suppress mTOR signaling promoted ER remodeling and depended on ER-phagy genes for lifespan extension, revealing a trade-off: preservation of ER mass conflicted with long-term survival under growth-restricted conditions.
To summarize, the new findings of Professor Kristopher Burkewitz and colleagues reposition ER morphology as an active variable in ageing biology and frames ER remodeling as a coordinated response that reallocates cellular capacity. Loss of rough ER reduces protein synthesis burden, while retention of tubular domains supports lipid handling and inter-organelle exchange. That shift aligns with known physiological demands of ageing cells, which favor maintenance over growth. The conservation of this pattern across yeast, worms, and mammals argues against species-specific pathology. It also reframes ER-phagy. Selective degradation of ER membranes appears less like emergency disposal and more like regulated downsizing. When ER-phagy fails, cells retain architecture mismatched to metabolic state, compromising survival under longevity-promoting conditions. We believe this perspective affects how stress pathways are interpreted. The unfolded protein response no longer functions solely as a reactive program but interfaces with physical remodeling that limits future stress exposure. Downscaling ER capacity early may reduce misfolding load later, even at the cost of reduced synthetic output. Such logic explains why long-lived states maintain low baseline UPR activity without sacrificing resilience. Design principles emerge from this view. Interventions that preserve youthful ER abundance may conflict with organismal lifespan, whereas strategies that support controlled remodeling could promote healthy ageing. The work also cautions against assuming that structural loss reflects damage. In some contexts, maintenance of excess ER may signal maladaptation rather than robustness. Future applications remain bounded. Manipulating ER-phagy carries risks, given tissue-specific requirements and dependence on intact autophagy networks. Yet the study provides a framework for evaluating ageing phenotypes through organelle architecture, offering a lens that integrates metabolism, proteostasis, and longevity without reducing ageing to molecular stress alone.
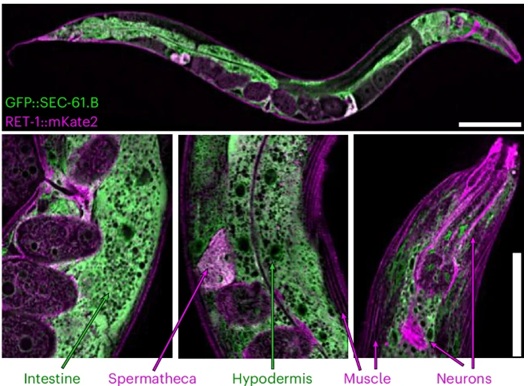
Figure legend: Differential enrichment of ER subdomain markers between tissues


Reference
Donahue EKF, Hepowit NL, Ruark EM, Mulligan AG, Keuchel B, Urban ND, Peng L, Stephens S, Johnson DJ, Wallace NS, Jackson LP, Ellisman MH, Arrojo E Drigo R, Folkmann AW, Truttmann MC, MacGurn JA, Burkewitz K. ER remodelling is a feature of ageing and depends on ER-phagy. Nat Cell Biol. 2026 . doi: 10.1038/s41556-025-01860-1.