
Significance
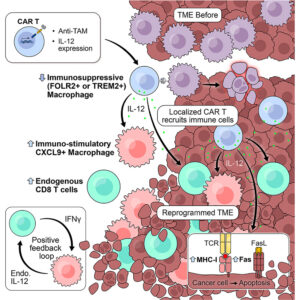
Tumors that grow despite a heavy immune infiltrate often fail to collapse because myeloid cells dampen cytotoxic pressure under inflammatory stress. In many solid cancers, macrophages that accumulate within lesions express markers such as FOLR2 or TREM2 and sustain a state in which T cells enter but do not act. This macrophage-dominated suppression persists even when cancer cells themselves present antigens capable of triggering recognition. CAR-T strategies aimed directly at malignant cells have struggled in this setting, partly because antigen loss or shared expression with healthy tissues constrains target choice, and partly because engineered T cells encounter a microenvironment already conditioned to blunt their activity. Efforts to remove macrophages wholesale have not translated well. Antibody-mediated depletion through CSF1R blockade erases broad macrophage pools, including populations required for tissue homeostasis, while leaving tumors able to reconstitute suppressive niches. More selective targeting of tumor-associated subsets has begun to address specificity, yet prior approaches that removed FOLR2-positive macrophages produced only transient control. Tumor growth resumed once the engineered cells contracted, implying that depletion alone does not impose a lasting change on the local immune state. Another layer of difficulty arises from cytokine biology. Factors such as interleukin-12 can drive potent anti-tumor immunity, but systemic exposure carries well-documented toxicity. Local delivery through engineered cells promises spatial restriction, though this strategy introduces new questions. How much cytokine can be released before toxicity emerges? Can localized cytokine production alter macrophage identity rather than simply eliminate them? And can such changes persist after the engineered cells diminish? These questions motivate a shift away from viewing macrophages only as obstacles to be removed. Tumor-associated macrophages display plastic transcriptional programs shaped by local cues. If a targeted intervention could both remove suppressive subsets and bias replacement populations toward immune-supportive states, the microenvironment itself might become an active participant in tumor control. Achieving this would require precise targeting, restrained cytokine output, and evidence that downstream immune circuits carry the therapeutic burden. A recent research paper published in Cancer Cell and conducted by Jaime Mateus-Tique, Ashwitha Lakshmi, Bhavya Singh, Rhea Iyer, Alfonso R. Sánchez-Paulete, Chiara Falcomatà, Matthew Lin, Gvantsa Pantsulaia, Alexander Tepper, Trung Nguyen, Angelo Amabile, Gurkan Mollaoglu, Luisanna Pia, Divya Chhamalwan, Jessica Le Berichel, Hunter Potak, Marco Colonna, Alessia Baccarini, Joshua Brody, Miriam Merad, and led by Professor Brian Brown from the Icahn School of Medicine at Mount Sinai in New York, The authors developed CAR-T cells that target FOLR2- or TREM2-expressing tumor-associated macrophages and deliver controlled interleukin-12 directly within tumors. They introduced a destabilization domain to tune cytokine output without altering CAR specificity. The system replaces suppressive macrophage populations with CXCL9-expressing macrophages and expands endogenous cytotoxic T cells.
Briefly, the research team first established that CAR-T cells directed against FOLR2 trafficked efficiently to tumor sites and selectively eliminated FOLR2-expressing macrophages without affecting macrophages lacking this marker. Investigators confirmed this behavior across ovarian and pancreatic tumor models, including settings where cancer cells themselves did not express the target antigen. Despite effective macrophage removal, tumor control remained limited, reinforcing the idea that depletion alone does not suffice. To convert these targeting vectors into delivery platforms, the authors engineered CAR-T constructs that secreted different cytokines upon engagement. The study compared interferons, interleukin-15 variants, and interleukin-12. In vitro, each armored construct retained macrophage-killing capacity and released biologically active payloads. In vivo testing revealed a sharper distinction. CAR-T cells releasing interferons failed to extend survival, while high-output interleukin-15 or interleukin-12 constructs induced severe toxicity at doses commonly used for CAR-T therapy. Excessive expansion of engineered T cells correlated with weight loss and early mortality, highlighting a narrow safety window. The investigators then adjusted cytokine exposure rather than abandoning the approach. Lowering cell dose and removing lymphodepletion reduced toxicity but also risked losing efficacy. To resolve this tension, the authors introduced a destabilization domain fused to interleukin-12, reducing steady-state cytokine levels while preserving inducible release during antigen engagement. This modification altered the balance decisively. At low doses and without preconditioning, interleukin-12 armored anti-FOLR2 CAR-T cells produced durable tumor regression with minimal systemic effects. Spatial transcriptomic analysis provided a mechanistic explanation. The researchers observed a sharp reduction in suppressive macrophage populations accompanied by expansion of CXCL9-expressing macrophages associated with T-cell recruitment. Endogenous CD8 T cells accumulated and displayed activation signatures that outlasted the presence of the engineered cells. Tumor clearance depended in part on FAS expression by cancer cells, linking cytokine-driven immune remodeling to a defined death pathway. Parallel experiments targeting TREM2-positive macrophages in lung metastasis models reproduced these patterns, suggesting that the effect was not limited to a single macrophage marker or tumor type. Across systems, the authors observed a recurring constraint: therapeutic benefit required tight control of cytokine dose and localization. Exceeding this boundary rapidly shifted benefit toward toxicity, a trade-off acknowledged rather than eliminated.
To summarize, the new work of Professor Brian Brown and colleagues reframes myeloid-directed immunotherapy from a subtractive maneuver into a reprogramming strategy and instead of treating macrophages solely as suppressive barriers, the study demonstrates that targeted cytokine delivery can reshape the composition and function of the macrophage compartment itself. The emergence of CXCL9-producing macrophages following treatment suggests that new immune-supportive niches arise rather than a simple void left by depletion. The findings also challenge assumptions about CAR-T cell persistence. Durable tumor control occurred even after engineered cells declined, implying that downstream immune circuits carried the response forward. That feature addresses a recurring limitation in solid tumor CAR-T efforts, where transient activity often parallels transient benefit. Here, macrophage reconditioning and endogenous T-cell expansion appear to extend the therapeutic footprint beyond the lifespan of the infused cells. Plus, the study highlights cytokine control as a central variable. Interleukin-12 retains powerful anti-tumor capacity when its release remains spatially confined and quantitatively restrained. The destabilization domain approach offers one way to modulate this balance, though translation will demand careful calibration across species and tumor contexts. The reliance on FAS-dependent killing also introduces a boundary condition: tumors lacking this pathway may respond differently. We believe the work establishes a framework for using myeloid targeting as an entry point to immune remodeling and if adapted cautiously, this strategy could complement existing T-cell-directed therapies or provide alternatives where tumor antigens remain elusive.

Reference
Mateus-Tique J, Lakshmi A, Singh B, Iyer R, Sánchez-Paulete AR, Falcomatà C, Lin M, Pantsulaia G, Tepper A, Nguyen T, Amabile A, Mollaoglu G, Pia L, Chhamalwan D, Le Berichel J, Potak H, Colonna M, Baccarini A, Brody J, Merad M, Brown BD. Armored macrophage-targeted CAR-T cells reset and reprogram the tumor microenvironment and control metastatic cancer growth. Cancer Cell. 2026 Jan 22:S1535-6108(25)00555-0. doi: 10.1016/j.ccell.2025.12.021.