
Significance
When glioma cells start expanding in the brain, they’re not just pushing against neurons and extracellular matrix. They run into microglia almost immediately. And that interaction isn’t straightforward. Depending on the context, microglia seem to restrain tumor growth or, in other reports, to support it. That inconsistency has complicated how we interpret tumor-associated macrophages in high-grade glioma. Clinical studies often correlate macrophage abundance with prognosis, but those analyses usually collapse distinct myeloid populations into one category. Resident microglia and infiltrating macrophages are developmentally different. Microglia derive from yolk sac progenitors and persist in the CNS as long-lived, self-renewing cells. Infiltrating macrophages come from circulating monocytes recruited during inflammation or tissue disruption. We tend to group them together under the TAM label, but their origins are not interchangeable, and neither are their transcriptional programs. While the M1/M2 framework is a convenient shorthand, it fails to capture the functional adaptability of microglia. These cells do not merely exist in static states; they actively phagocytose debris, release cytokines, and interface with T cells in a manner that can alternately inhibit or facilitate tumor growth and angiogenesis. Those findings aren’t necessarily contradictory. They likely reflect differences in tumor stage, local cell density, or immune competence. What’s been missing are direct comparisons between resident microglia and infiltrating macrophages under controlled conditions. Much of what we assume about microglia has been extrapolated from peripheral macrophage biology, and that extrapolation may be misleading.
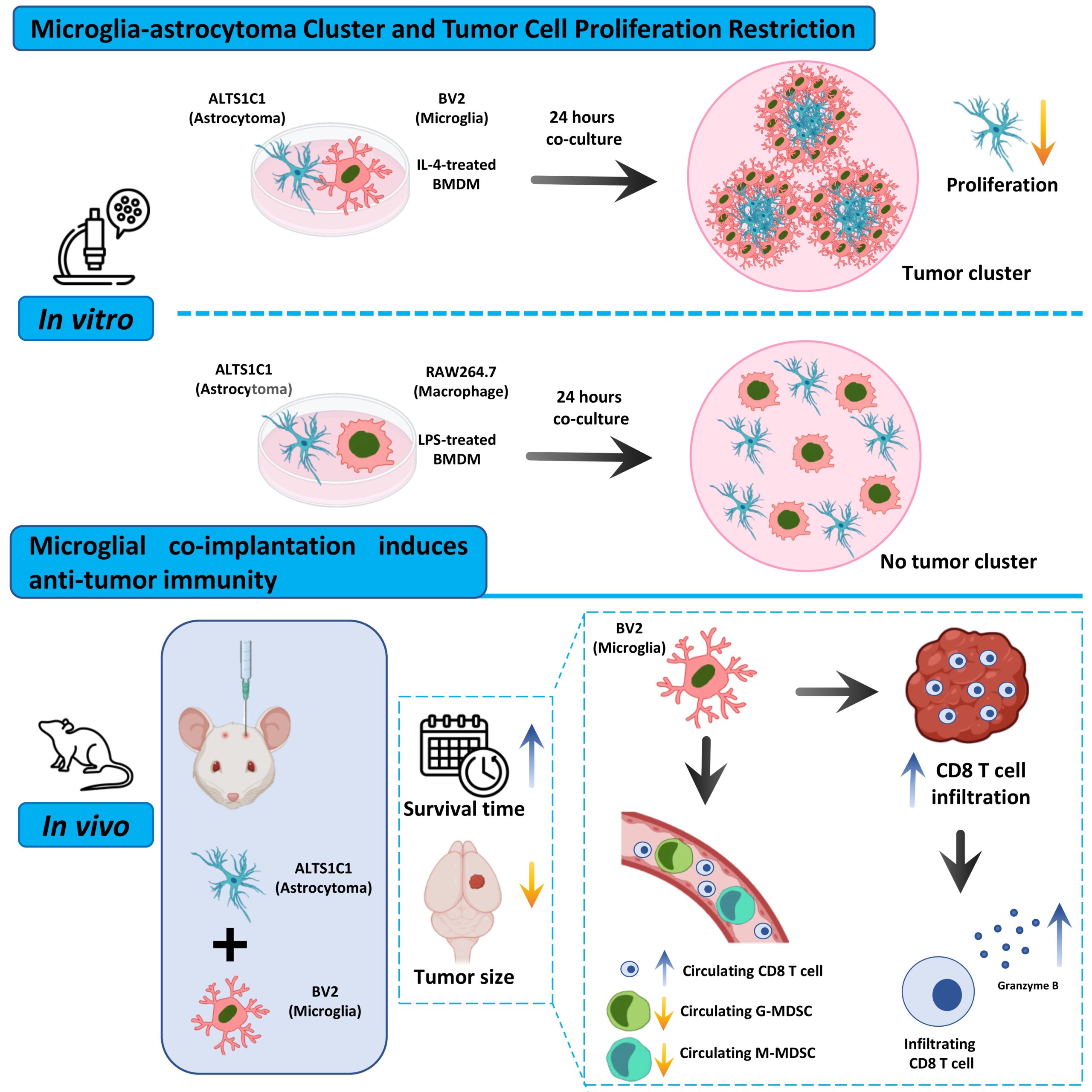
There’s also the unresolved question of how these myeloid populations influence CD8⁺ T-cell infiltration. In aggressive glioma, cytotoxic T cells are often sparse. If microglia alter tumor organization or proliferation kinetics, they may indirectly influence antigen exposure or chemokine gradients, and therefore T-cell access. Testing that idea requires separating these cell types experimentally and evaluating tumor growth in both immune-competent and T-cell-deficient settings. Without that separation, we’re still guessing. A recent research paper published in Journal of Molecular Oncology and conducted by Tzu-Chieh Sun, Ching-Fang Yu, Sheng-Yan Wu, Wei-Chung Cheng, Chi-Shiun Chiang, and led by Professor Fang-Hsin Chen from the National Tsing-Hua University, the researchers developed a density-controlled coculture platform that distinguishes microglial from macrophage effects on glioma organization and proliferation. They combined this system with orthotopic co-implantation models in immunocompetent and T-cell-deficient mice to dissect immune dependence.
Briefly, the research team established coculture systems pairing the murine astrocytoma line ALTS1C1 with either BV2 microglia or RAW264.7 macrophages at defined ratios. When they seeded BV2 together with ALTS1C1 at intermediate ratios, they observed discrete tumor cell clusters exceeding 50 μm in diameter. Time-lapse imaging revealed that BV2 actively drove tumor cells toward one another, concentrating them into compact aggregates. Immunofluorescence showed ALTS1C1-GFP cells occupying the cluster core, while CD11b-positive BV2 cells formed a peripheral ring. When the investigators reduced either cell population below a threshold density, clustering failed to occur, indicating that spatial confinement required sufficient microglial presence.
In contrast, the authors performed identical cocultures with RAW264.7 cells and detected no cluster formation at any ratio. Tumor and macrophage cells distributed homogeneously, and dynamic imaging didn’t reveal coordinated aggregation. They extended this comparison using bone-marrow-derived macrophages polarized with IL-4 or LPS. Only IL-4–treated macrophages recapitulated the clustering phenotype, whereas LPS-treated cells didn’t. Bulk RNA sequencing showed that BV2 expressed both M1- and M2-associated transcripts at higher levels than RAW264.7, reflecting a transcriptional state not easily reduced to a single polarization category; that composite signature may enable behaviors absent in the peripheral macrophage line.
To probe the proliferative consequences of clustering, the investigators conducted cell cycle analysis on ALTS1C1-GFP after coculture with BV2 and documented a reduction in G0/G1 and S-phase fractions and a marked increase in G2/M-phase cells, consistent with growth restraint accompanied by cell cycle arrest. BV2 proliferation did not decrease under the same conditions, which argues against simple nutrient competition and imply that microglial confinement alters tumor cell cycle progression. The team then examined orthotopic implantation in immunocompetent C57BL/6 mice. Mice receiving ALTS1C1 alone exhibited a mean survival of approximately 30 days. When the researchers co-injected equal numbers of BV2 and tumor cells, survival extended beyond 77 days, and tumors were undetectable at that endpoint. Reducing BV2 numbers attenuated, but did not abolish, the survival benefit. Histological analysis at days 10 and 24 showed smaller tumors in BV2-containing groups, with a number-dependent effect. Interestingly, in the highest BV2 ratio group, tumor size at day 24 did not exceed that at day 10, suggesting that mechanisms beyond initial proliferation arrest contributed to delayed progression.
To test the contribution of adaptive immunity, the investigators repeated implantation in T-cell-deficient SCID mice. In this setting, the survival advantage conferred by BV2 diminished substantially, and by day 24 tumors expanded more aggressively than in immunocompetent hosts. Immunofluorescence staining revealed increased infiltration of CD8⁺ T cells and elevated Granzyme B expression in BV2-containing tumors, especially at later time points. Peripheral blood analysis demonstrated preservation of circulating CD8⁺ T-cell levels and suppression of granulocytic and monocytic myeloid-derived suppressor cell expansion in BV2-containing tumor-bearing mice. The protective effect thus correlated with sustained cytotoxic T-cell presence and reduced systemic immunosuppression. Still, the data leave open whether microglia directly instruct T cells or alter tumor architecture in a way that secondarily facilitates immune access.
The findings of Professor Fang-Hsin Chen and colleagues argue against the assumption that myeloid cells uniformly drive glioma progression and by separating resident microglia from infiltrating macrophages, the study identifies a lineage-dependent behavior that often gets lost in aggregate analyses. Microglia do not simply accompany tumor growth; under defined conditions, they reorganize tumor cells spatially and constrain proliferation while coinciding with increased cytotoxic T-cell activity. If developmental origin shapes functional capacity, then therapeutic strategies that broadly eliminate “macrophages” risk removing cell populations that may contribute to tumor control in certain contexts. The clustering phenomenon deserves closer consideration. Physical confinement of tumor cells alters cell–cell contact and likely perturbs diffusion gradients within the lesion. The observed G2/M accumulation suggests that microglial proximity interferes with orderly cell cycle progression, whether through mechanical constraint or local signaling. The sequence that emerges is not linear but conditional: microglial density reshapes tumor architecture; altered architecture associates with reduced proliferation and greater CD8⁺ T-cell presence; immune competence ultimately determines whether this configuration produces sustained control. In T-cell-deficient hosts, structural restraint alone does not prevent eventual tumor expansion, which places adaptive immunity as a necessary amplifier instead of a bystander. The systemic findings extend this picture and BV2 co-implantation correlates with restrained expansion of circulating MDSCs and preservation of CD8⁺ T-cell levels, implying that local myeloid–tumor interactions reverberate beyond the brain microenvironment. That broader immune effect, suggests that resident microglia may influence both spatial tumor organization and systemic immune balance.

Reference
Sun TC, Yu CF, Wu SY, Cheng WC, Chiang CS, Chen FH. Microglia limit brain tumor development by restricting tumor cell proliferation and inducing T-cell immunity. Mol Oncol. 2025;19(9):2670-2685. doi: 10.1002/1878-0261.70102.
Go to Journal of Molecular Oncology.