
Significance
When chromatin loops must be rebuilt as cells leave mitosis, any interruption in cohesin activation can alter the contact architecture that genes encounter as transcription resumes. That problem sits near the center of current thinking about three-dimensional genome control, because loop extrusion is now treated as a major organizing process for subchromosomal structure, yet its kinetics inside mammalian cells have remained hard to define directly. Live-cell work had already implied short-lived loops, and biochemical work had established NIPBL as a key accessory factor for cohesin. Even so, a basic question stayed unsettled: does continuous NIPBL activity simply keep loops running at all loci, or does its importance depend on when a loop is being formed and what kind of chromatin the loop traverses?
A second uncertainty followed from transcription. Chromatin architecture has long been linked to gene control, but acute disruption of genome folding often changes expression far less than one might expect from the scale of structural disturbance. That mismatch left the field with an unsatisfying picture. Either many contacts were more dispensable than assumed, or only a particular subset of genes depended on a specific form of cohesin behavior that standard perturbations had not isolated cleanly. In a recent research paper published in Nature Genetics, Dr. Tessa Popay, Ami Pant, Dr. Femke Munting, Dr. Melodi Tastemel, Dr. Morgan Black, Dr. Nicholas Haghani &led by Professor Jesse Dixon from the Salk Institute of Biological Studies, developed an acute degron-based system for NIPBL depletion in human cells and used it to separate ongoing loop establishment from persistence of pre-existing chromatin contacts. They combined that perturbation with Hi-C, ChIP-seq, ChromHMM, and nascent-transcription profiling during both asynchronous growth and mitotic exit. The technical distinction from prior cohesin-removal studies is that this framework interrogates NIPBL-dependent loop extrusion dynamics directly rather than collapsing all cohesin-associated architecture at once. They also extended the same strategy to hiPSC-derived neurons and cardiomyocytes, allowing the local structural signature of NIPBL-sensitive genes to be compared across lineages.
The study was motivated as much by timing as by mechanism. Mitotic exit provides a natural reassembly window for genome structure, and that window makes it possible to separate loop formation from loop persistence in a living mammalian system. If a contact disappears only when NIPBL is removed during post-mitotic reorganization, but remains detectable when NIPBL is depleted in asynchronous cells, then the locus is revealing something important about maintenance logic rather than mere presence or absence of cohesin. The same reasoning extends to transcriptional activation after mitosis. Genes re-entering expression do not do so against a static chromatin background; they do so while local contact patterns are being rebuilt. The central problem, then, was not simply whether NIPBL affects chromatin loops or whether it affects RNA output. The deeper issue was whether a defined mode of loop extrusion produces a local genomic arrangement that certain genes require as they regain or sustain their active state.
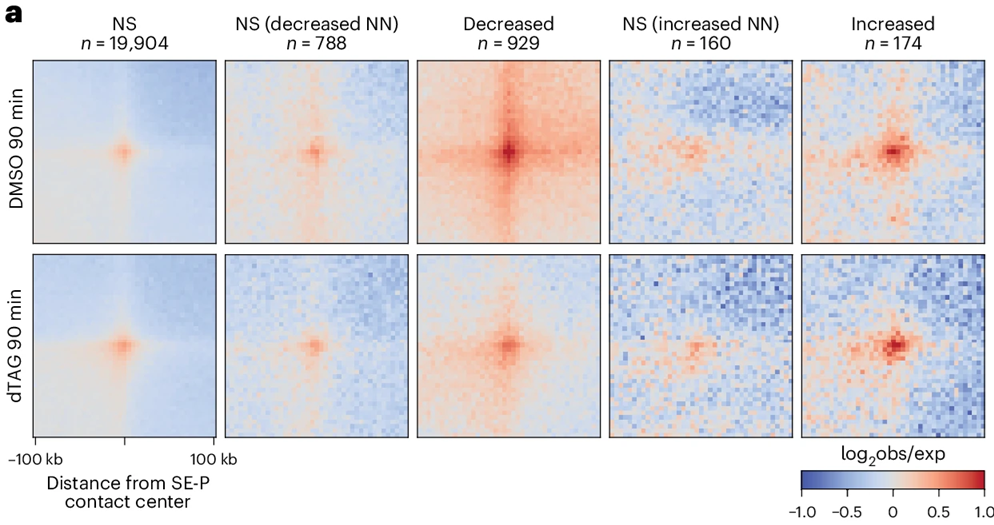
The researchers first engineered hTERT RPE-1 cells for rapid dTAG-mediated depletion of NIPBL and paired that system with RAD21 depletion so they could distinguish loss of loop establishment from loss of cohesin itself. NIPBL removal began quickly, MAU2 fell with it, and RAD21 binding decreased across chromatin even though bulk chromatin-associated cohesin dropped only modestly. That difference is informative rather than incidental: a cell can retain chromatin-associated cohesin in a broad biochemical sense while still losing the locus-specific engagements that generate recognizable loops. Using Hi-C and RAD21-based classification, they defined 16,860 cohesin-dependent loops and found that these loops did not respond uniformly. Some declined sharply after NIPBL depletion, whereas another class persisted for hours. The mixed-dependency group became especially interesting because it weakened more at 24 hours than at 4 hours, implying that persistence reflected longevity of pre-existing structures rather than continued renewal. They then used mitotic exit as a direct test of formation logic. After synchronizing cells in mitosis, depleting NIPBL, and following genome reorganization through early G1, they saw that all loop classes depended much more strongly on NIPBL during reassembly than they did in asynchronous cells. The same mixed-dependency loops that could linger after acute depletion in cycling cells failed to establish properly during mitotic exit. It argues that persistence and establishment are separable properties in vivo, and it explains why an acute perturbation of NIPBL can expose loop lifetimes that would be invisible if one removed cohesin altogether.
The team pushed further by asking what marks the loops that endure. STAG1 enriched at the more NIPBL-dependent and persistent contacts, STAG2 showed the opposite pattern, and STAG1 knockout specifically weakened the mixed-dependency class after NIPBL depletion, whereas STAG2 knockout strengthened it. That pattern fits a residence-time model in which STAG1-containing cohesin remains chromatin-bound longer and can preserve selected contacts after new extrusion events have been curtailed. Chromatin context also tracked with loop behavior. Less dependent loops more often involved enhancer-rich regulatory elements, while the mixed-dependency class carried a stronger association with repressive chromatin and H3K27me3-flanked anchor environments. The locus mattered because loop stability was not being dictated by one universal timer; it was being shaped by subunit composition, chromatin state, and genomic setting together.
To connect structure with gene control, the researchers measured nascent transcription during mitotic exit by SLAM-seq. NIPBL depletion altered 549 genes in RPE-1 cells, with 457 reduced and 92 increased. Genes that failed to activate were enriched for programs tied to cell migration, cell shape, epithelial-mesenchymal transition, and KRAS signaling, and prolonged depletion shifted cell morphology toward a more epithelial-like state. Kinetic measurements sharpened the interpretation: reduced genes still began activation at roughly the normal early interval, yet they failed to build to full output after 60 minutes. That timing argues that NIPBL-dependent genome reorganization does not merely trigger the first burst of transcription; it supports continued productive activation as cells progress into G1. The spatial analysis matched that idea. Sensitive genes showed stronger contact with nearby super-enhancers and typical enhancers, along with weaker insulation across the transcription start site and stripe-like contact patterns extending through the promoter region. When NIPBL was removed, those local configurations weakened. In hiPSC-derived neurons and cardiomyocytes, the same design logic reappeared in a lineage-specific form: most dysregulated genes were cell-type-specific, yet the affected genes again displayed stronger enhancer proximity and characteristic promoter-centered contact structures.
Professor Jesse Dixon and his team demonstrated that NIPBL-sensitive genes are embedded in a local contact geometry marked by close association with neighborhood super-enhancers, permissive communication across the transcription start site, and coordinated engagement among multiple regulatory elements. That arrangement gives cohesin dynamics a specific regulatory meaning. Rather than viewing loop extrusion as a broad architectural background process, the study places it inside the local operating logic of selected cell-identity genes. Genes of that class appear to need an active contact regime that can sustain transcriptional build-up, especially during post-mitotic reactivation. The study also links persistence to composition. STAG1 dependence and chromatin-state association are not decorative annotations on the loop classes. They provide a molecular explanation for why genome folding does not decay uniformly when NIPBL is removed. That matters for future work on cohesin regulation, because it means that the effect of perturbing a loader or ATPase regulator cannot be inferred simply from the effect of removing a core ring component. Distinct perturbations interrogate distinct layers of genome organization. Acute NIPBL depletion is especially useful in that respect, since it reveals how contact renewal shapes living chromatin without immediately erasing every cohesin-dependent feature. The cross-lineage data extend the argument without overstating it. Neurons and cardiomyocytes did not merely recapitulate the RPE-1 response; they did so in a cell-type-specific transcriptional register, with different gene sets but a shared structural signature. That convergence supports a general design principle inside the scope of the experiments: NIPBL supports expression of lineage-defining genes by organizing a local promoter-enhancer neighborhood with unusually strong spatial coupling. The new study also provides a more exact way to think about why architectural perturbations often affect only a subset of genes. Sensitivity may depend less on promoter class alone and more on whether a gene relies on this particular contact topology to reach full transcriptional output.


Reference
Popay TM, Pant A, Munting F, Tastemel M, Black ME, Haghani N, Dixon JR. Acute NIPBL depletion reveals in vivo dynamics of loop extrusion and its role in transcription activation. Nat Genet. 2026 Feb 16. doi: 10.1038/s41588-026-02516-y.
Go to Nature Genetics