
Significance
Tumor control fails to follow the usual immunological script when colorectal cancer accumulates large numbers of Foxp3-positive regulatory T cells yet does not behave like other solid tumors in which Treg enrichment tracks with tumor support. That mismatch is the starting problem here. In most cancer settings, abundant intratumoral Treg cells accompany suppression of anti-tumor immunity and poor outcome. Colorectal cancer, especially the microsatellite-stable form that carries proficient mismatch repair and resists combined PD-1 and CTLA-4 blockade, does not fit comfortably into that rule. In a recent research paper published in Immunity Journal and led by Professor Alexander Rudensky from the Memorial Sloan Kettering Cancer Center, developed a functional framework that divides colorectal cancer Treg cells into IL-10-positive and IL-10-negative subsets with opposite effects on tumor growth. They paired orthotopic AKP colorectal cancer organoids, single-cell RNA/ATAC profiling, fate mapping, and reciprocal genetic depletion systems to test those subsets directly. They also established a mechanistic model in which Treg-derived IL-10 restrains CD4-positive-cell IL-17 production and, through that route, limits IL-17RA-dependent proliferation of tumor cells. Human multiomic and clinical analyses extended the same framework to patient colorectal cancer and linked the two Treg states to opposite prognostic associations.
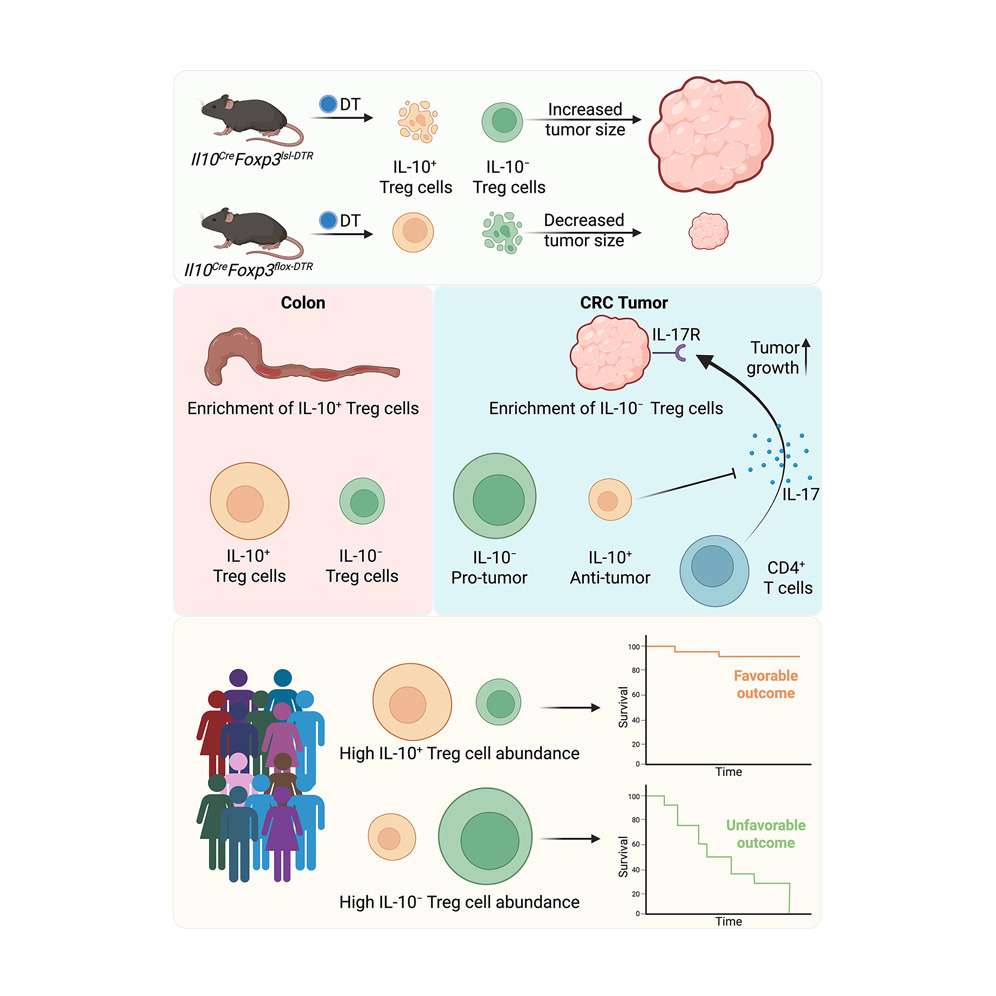
If Treg cells are treated as one population, then their enrichment in colorectal cancer becomes hard to reconcile with the association between Treg and CD8-positive T cell density and improved tumor control in human disease. Pan-Treg depletion in other tumor models often restrains cancer growth, yet the same maneuver would not necessarily clarify colorectal cancer if distinct Treg states were exerting different forms of control at the same time. The unresolved issue was not simply whether Treg cells matter in colorectal cancer, but whether separate Treg programs coexist there and redirect immunity in opposite directions. Earlier work on colonic Treg heterogeneity had already shown differences in ontogeny, differentiation, and effector properties, including a subset marked by IL-10 expression with recognizable tissue-protective activity in the colon. That prior knowledge made it plausible that one fraction of tumoral Treg cells might restrain tumor-supporting inflammation even as another fraction preserves the familiar suppressive pattern seen in many cancers.
The researchers approached that problem by choosing an orthotopic mouse model built from Apc loss, Trp53 loss, and oncogenic KrasG12D organoids, because the model reproduces several defining properties of human microsatellite-stable colorectal cancer: luminal tumor growth, spontaneous spread to mesenteric lymph nodes and liver, a microenvironment poor in effector CD8-positive activity, enriched in Treg cells and macrophages, and resistant to PD-1 blockade. That choice matters conceptually. A model with the wrong immune architecture would flatten the very heterogeneity under question, whereas a model that preserves the immunosuppressive texture of human MSS colorectal cancer gives the Treg compartment room to separate into functionally meaningful states. Against that background, the authors set out to compare tumoral and adjacent colonic Treg cells at single-cell resolution in mice and humans, track how these populations change during tumor progression, and test whether a particular cytokine-defined split inside the Treg pool could explain why colorectal cancer behaves so differently from the dominant pattern across solid tumors. The first decisive observation came from single-cell RNA and ATAC profiling of T cells from AKP tumors and adjacent cecal tissue. Rather than revealing one activated Treg compartment, the analysis separated Treg cells into two clusters distinguished by Il10 expression. IL-10-positive Treg cells were more common in normal cecum, whereas IL-10-negative Treg cells accumulated in tumors. A time-course analysis across 2, 4, and 6 weeks sharpened that picture: tumors progressively enriched the IL-10-negative subset without reducing the frequency of the IL-10-positive subset, and this shift unfolded alongside changes in effector populations such as PD-1-positive Th1 cells and early Pdcd1-high CD8-positive cells. That pattern matters because it frames the tumor not as a site of general Treg accumulation, but as an environment that selectively favors one Treg state over another. The authors then used fate-mapping strategies to show that the tumoral Treg pool largely arose from migration and expansion of preexisting Treg cells, and that preexisting IL-10-positive Treg cells largely retained their IL-10-positive identity.
Single-cell chromatin and transcriptomic analysis then gave each subset a molecular identity. IL-10-positive Treg cells were associated with Rorc, Maf, Ctla4, Ccr2, Zeb2, Gzmb, and Il23r, whereas IL-10-negative Treg cells preferentially expressed Ikzf2, Il1rl1, Gata3, Klrg1, Zbtb46, Itgb8, and Il2ra. Differential enhancer accessibility at the Il10 and Ikzf2 loci paralleled those transcriptional differences, which is important because it places the split at the level of gene-regulatory circuitry rather than transient cytokine staining alone. The IL-10-negative program also carried selective enrichment for NR4A-family motif activity and the strongest TCR-signaling-related gene score among the compared subsets, fitting a state of heightened activation inside tumors. That logic helps explain why the tumor environment would accumulate IL-10-negative cells: the local antigenic and inflammatory conditions appear to reinforce a program tuned to strong receptor engagement.
Functional testing came from elegant reciprocal depletion systems. Selective ablation of IL-10-positive Treg cells enlarged tumors, whereas depletion of IL-10-negative Treg cells reduced tumor size and was accompanied by stronger CD8-positive responses and heavy mononuclear infiltration. The two perturbations also changed the immune composition in opposite directions. Removing IL-10-positive Treg cells increased monocytes, macrophages, neutrophils, Spp1-positive tumor-associated macrophages, and a CD4 program rich in Rorc, IL-17, and IL-22, with reduced IL-4 and IL-13. Removing IL-10-negative Treg cells produced the reverse immune tilt, with more IFNγ-producing CD8-positive cells and Th2 cytokines and fewer IL-17-expressing Th17 cells. The mechanistic core of the paper rests on the IL-10–IL-17 axis. IL-17, not IL-22, accelerated growth of AKP organoids in culture. AKP tumors lacking IL-17RA were smaller in vivo, which places tumor-cell IL-17 sensing directly in the causal chain. Blocking IL-10Rα increased CD4-positive IL-17 production and tumor size in a way that mirrored IL-10-positive Treg depletion, and IL-17RA-deficient tumors no longer responded to either IL-10R blockade or IL-10-positive Treg depletion. Chimeric experiments deleting Il10 specifically in Treg cells reached the same conclusion from the ligand side: Treg-derived IL-10 restrains tumor burden through a pathway that depends on tumor IL-17RA. That sequence of experiments works because each intervention removes a different link in the same circuit, and the circuit remains coherent every time it is tested. Human material aligned with the mouse work: patient-derived tumor organoids responded to recombinant IL-17 with faster growth, and paired human tumor and adjacent tissue multiomics recovered IL10-positive RORC-linked and IL10-negative IKZF2-linked Treg subsets with the same tissue bias seen in mice.
Professor Alexander Rudensky and colleagues demonstrated that one Treg subset can restrain tumor growth because it keeps a tumor-promoting IL-17 program in check, whereas another subset supports progression by suppressing CD8-positive and type 2 responses. That reframing matters well beyond nomenclature. It replaces a quantity-based view of Treg biology with a composition-based view, and once that shift is made, the long-standing colorectal exception becomes much easier to understand. Moreover, broad depletion of all Treg cells in the AKP model left tumor growth largely unchanged, yet selective depletion of one subset or the other produced strong and opposite outcomes. The paper makes a persuasive point that mixed cell populations can conceal real biology through cancellation. In practical terms, therapeutic strategies aimed at “Treg targeting” in colorectal cancer cannot be judged only by whether they reduce Treg abundance. Their effect will depend on which Treg state is being removed, preserved, or shifted. The authors connect that reasoning to PD-1 resistance in MSS colorectal cancer by linking the pro-tumoral IL-10-negative subset to strong activation features and elevated Pdcd1 expression. That gives the work translational relevance without leaving the evidence base provided in the paper. Plus, in patient samples, IL-10-positive and IL-10-negative Treg cells showed opposite tissue distributions in normal adjacent colon and tumor, matched the mouse subsets in chromatin and transcriptional features, and tracked with better and worse prognosis, respectively. That agreement matters because the paper is not asking readers to extrapolate from an isolated mouse phenotype to human disease. It shows that the same internal split appears in human colorectal cancer and that the split carries clinical meaning. The extension into published pan-cancer single-cell atlases makes the conceptual reach a bit wider: related IL-10-positive and IL-10-negative Treg-like states appeared in several barrier tissue cancers, including colorectal, basal cell, head and neck, and stomach tumors. The implication is measured but important. The circuitry defined in colorectal cancer may belong to a broader class of tissue-shaped immune regulation in barrier malignancies.
Indeed, the work reorients how we think about immune restraint inside tumors that develop in chronically exposed barrier tissues. IL-10 from Treg cells, in this setting, is not simply a general anti-inflammatory output. It becomes part of a specific protective circuit that prevents CD4-positive cells from feeding tumor growth through IL-17. Once that relationship is recognized, selective targeting of the IL-10-negative subset becomes a more coherent design principle than undifferentiated Treg depletion. The field has often had to choose between preserving Treg biology for tissue stability and removing it for anti-tumor immunity. This study shows that, at least in MSS colorectal cancer, the choice can be posed more precisely.


Reference
Xiao Huang, Dan Feng, Sneha Mitra, Emma S. Andretta, Nima B. Hooshdaran, Aazam P. Ghelani, Eric Y. Wang, Joe N. Frost, Victoria R. Lawless, Aparna Vancheswaran, Qingwen Jiang, Cheryl Mai, Karuna Ganesh, Christina S. Leslie, Alexander Y. Rudensky. Opposing functions of distinct regulatory T cell subsets in colorectal cancer. Immunity, 2026; 59 (1): 145 DOI: 10.1016/j.immuni.2025.11.014
Go to Immunity