
Significance
Protein–protein interactions often resist modulation by conventional small molecules because their interfaces extend across broad and relatively shallow surfaces that lack deep binding pockets. Under such geometric conditions, ligand frameworks that depend on rigid scaffolds or small contact areas struggle to achieve sufficient affinity or specificity. Cyclic peptides partially overcome this limitation because backbone cyclization restricts conformational freedom while preserving a flexible interface capable of contacting large protein surfaces. Structural constraint reduces entropic penalties upon binding and frequently stabilizes conformations compatible with recognition of extended protein interfaces. For this reason, cyclic peptides continue to attract attention as candidates for therapeutic strategies directed at protein interaction networks that resist classical drug design approaches. However, still practical design of cyclic peptide binders presents a methodological difficulty. The combinatorial space defined by peptide length, sequence composition, backbone conformation, and cyclization geometry expands rapidly even for modest ring sizes. Conventional computational pipelines struggle to explore this space efficiently. Fragment assembly strategies may provide a partial solution because they allow peptide growth from structural motifs derived from protein interfaces, but they require effective guidance to avoid enormous sampling overhead. Structural data availability also imposes limitations. Only a modest number of protein–cyclic peptide complexes have been solved experimentally, restricting the amount of direct training data available for structure-based generative approaches. Machine learning methods have already reshaped several areas of molecular design, including small molecules and protein structure prediction. Attempts to apply similar concepts to cyclic peptide construction remain comparatively sparse. Existing strategies frequently rely on backbone matching procedures or template extension, and these approaches often struggle when the design objective requires generation of novel cyclic conformations rather than modification of existing scaffolds and the challenge becomes even more acute when the desired ligand must bind a protein interface that lacks a known cyclic peptide template.
Designing peptides directly within the geometry of a protein binding site offers a more ambitious alternative. In such a strategy, fragments derived from known interfacial structures supply physically realistic building blocks while a search algorithm determines how these fragments combine into closed peptide rings. The conceptual motivation behind the present study arises from that possibility. If fragment growth could be guided adaptively by an algorithm capable of learning favorable assembly decisions during the search process, the exploration of cyclic peptide space might become both faster and structurally meaningful. Such reasoning motivated the development of a reinforcement-learning-driven framework intended to construct cyclic peptide binders directly on protein surfaces while accounting simultaneously for binding affinity, structural stability, and ring closure feasibility.
A recent research paper is published in Journal of Medicine Chemistry and conducted by Dr. Fanhao Wang, Mr. Jintao Zhu, Professor Changsheng Zhang, and Professor Luhua Lai from Peking University working together with Ms. Tiantian Zhang and Professor Xiaoling Zhang from Zhengzhou University, the researchers developed CYC_BUILDER, a computational framework that constructs cyclic peptide binders through fragment assembly guided by reinforcement learning and Monte Carlo Tree Search. The system grows peptides directly within protein binding interfaces while evaluating binding energy, structural stability, and cyclization feasibility. A large fragment database derived from protein–protein interface motifs supplies realistic building blocks for peptide growth. The authors created the fragment database by extracting interface motifs from experimentally determined protein complexes. Over one million tripeptide fragments and hundreds of thousands of tetrapeptide fragments were collected from structural datasets after filtering for redundancy and geometric consistency. The team classified fragments according to residue properties and backbone conformations to reduce sampling complexity during the search process. Fragment fusion algorithms then splice candidate motifs onto the growing peptide backbone while maintaining realistic geometry.
At each step of peptide growth, the algorithm evaluates candidate structures through a composite scoring function. The scoring model includes energetic terms associated with peptide–protein interaction energy, peptide structural stability, interface complementarity, and cyclization feasibility. The investigators used these values as rewards in the reinforcement learning procedure, allowing the search algorithm to favor fragment choices that produce promising intermediate structures. The design process continues until a termination condition triggers cyclization through either head-to-tail amide closure or disulfide bond formation.
The research group first examined whether the method could reconstruct known cyclic peptide binding modes. They assembled a benchmark dataset of nineteen protein complexes containing cyclic peptide ligands ranging from six to twenty residues. The algorithm generated new peptide candidates for each target structure while restricting cyclization chemistry to match the native ligand type. Many generated peptides reproduced key binding contacts found in experimental complexes, and predicted binding energies frequently matched or exceeded those of native ligands. Docking tests further indicated that the generated peptides adopted stable binding conformations, with most predicted poses remaining within a few angstroms of the reference structures.
Afterward, the investigators explored de novo ligand discovery against tumor necrosis factor alpha (TNFα), a cytokine involved in inflammatory signaling. The design procedure generated one hundred thousand cyclic peptide candidates targeting the protein surface. Filtering stages relied on structural energy criteria, molecular dynamics simulations, and free energy calculations. Nine peptides advanced to experimental testing. The authors demonstrated using surface plasmon resonance experiments measurable binding for several candidates, including one peptide with micromolar affinity. Additional cellular assays demonstrated that the selected peptides inhibited TNFα-mediated signaling responses, consistent with disruption of the TNFα–TNFR interaction pathway. The research team also observed a practical limitation embedded within the filtering strategy. Molecular dynamics simulations lasting one hundred nanoseconds sometimes failed to capture relevant conformational states, causing experimentally active sequences to rank lower in computational screening. That observation reflects a general trade-off in simulation-driven design workflows: deeper sampling improves structural reliability but substantially increases computational cost.
To summarize, cyclic peptides occupy a strategic niche in molecular therapeutics because they combine several properties typically distributed across different ligand classes. Their ring topology constrains backbone geometry while retaining enough flexibility to adapt to extended protein surfaces. Such structural characteristics allow cyclic peptides to engage targets that evade both small molecules and antibodies. Protein–protein interfaces involved in inflammatory signaling, immune regulation, and oncogenic pathways frequently fall into this category. The framework introduced by Professor Luhua Lai and colleagues demonstrates that reinforcement learning can guide structural assembly processes during peptide design. Instead of enumerating peptide sequences blindly, the algorithm modifies its sampling strategy dynamically according to intermediate structural evaluations. This adaptive search behavior addresses one of the persistent obstacles in peptide design: the overwhelming size of the sequence–structure space. When the algorithm identifies fragment combinations that stabilize peptide conformations within the binding interface, it biases subsequent exploration toward related structural motifs. Over time the search concentrates around conformations that balance binding strength, structural feasibility, and ring closure geometry. Another implication concerns structural diversity. Plus, the generated peptide exhibited substantial variation in backbone geometry and sequence composition, which indicate that the new algorithm doesn’t produce a narrow set of solutions and this is important because diversity is critical in drug discovery and different scaffolds can have distinct pharmacokinetic properties or resistance profiles which will maximize the chance of drug candidate to be successful medicine.
Computational efficiency also shapes the potential utility of this approach. Fragment-guided reinforcement learning allows the generation process to run on modest computing resources while maintaining reasonable search depth. The comparison against existing cyclic peptide design approaches shows that the method produces competitive binding energies while requiring far less runtime. This efficiency matters when screening large peptide libraries against multiple targets. At the same time, the experimental validation results illustrate the gap that still separates computational predictions from biochemical success. Only a subset of candidates identified through computational screening produced measurable activity in biochemical assays. Simulation time scales and scoring models inevitably simplify the physical behavior of peptides interacting with proteins in solution. Future development could incorporate improved conformational sampling strategies or refined energy models to better capture rare but functionally relevant conformations. Overall, the authors’ algorithm CYC_BUILDER and the de novo designed cyclic peptides that can disrupt TNFα signaling illustrate the broader potential of algorithm-guided peptide generation and similar strategies could address other protein interaction interfaces that resist conventional ligand design.
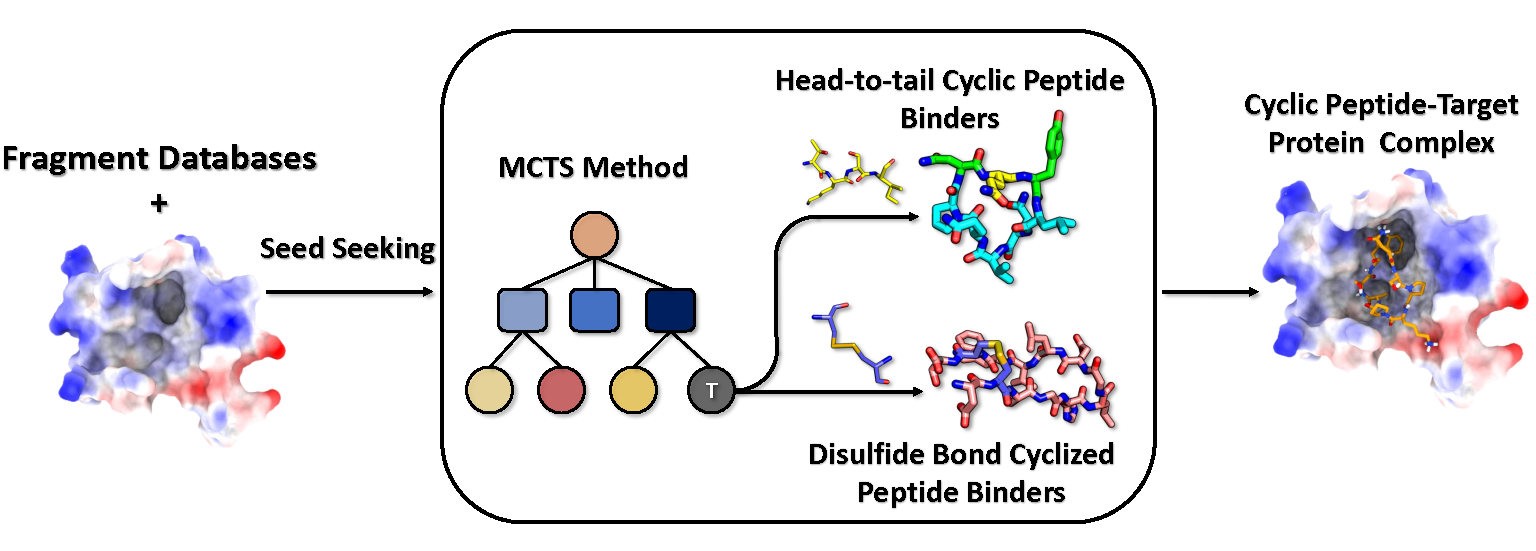
Figure 1. The overall scheme of CYC_BUILDER
Reference
Wang F, Zhang T, Zhu J, Zhang X, Zhang C, Lai L. Reinforcement Learning-Based Target-Specific De Novo Design of Cyclic Peptide Binders. J Med Chem. 2025;68(16):17287-17302. doi: 10.1021/acs.jmedchem.5c00789.
Go to Journal of Medicinal Chemistry