
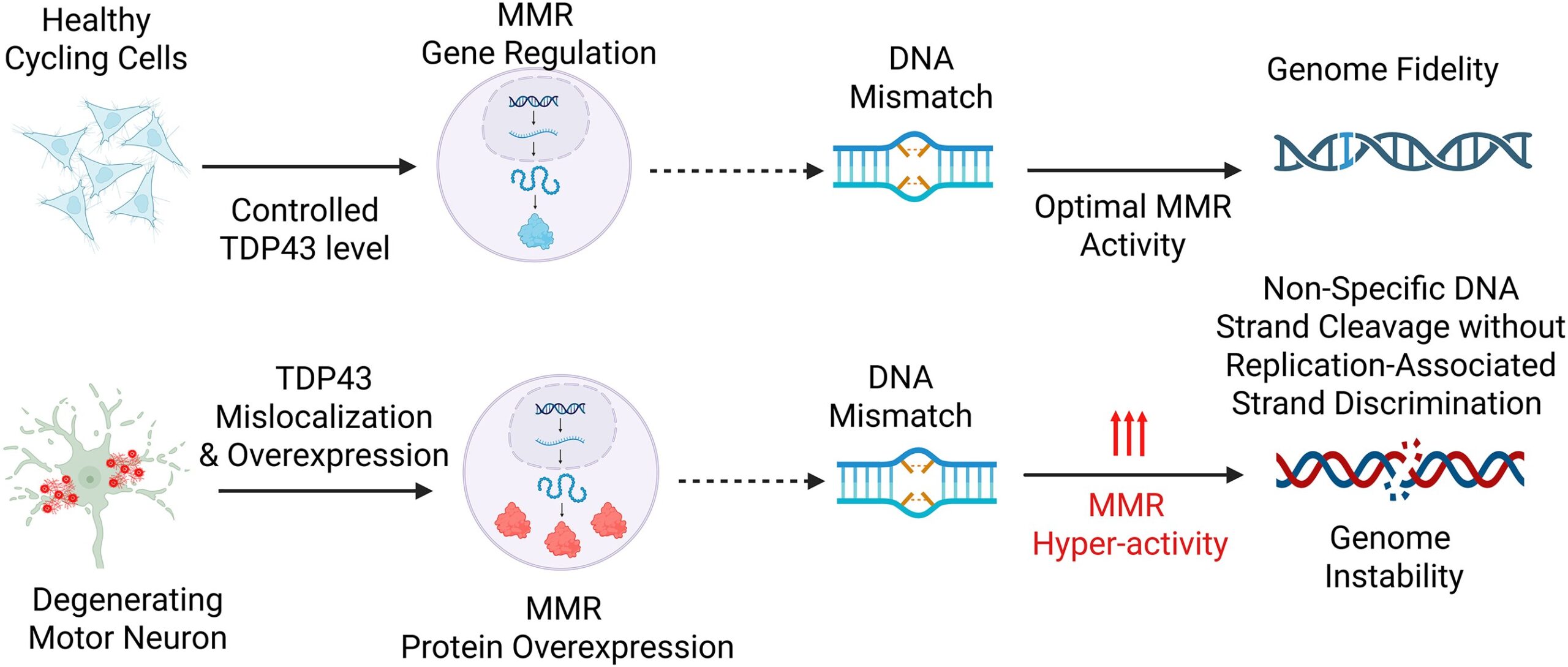
Significance
DNA mismatches become hazardous when the proteins that recognize and process them drift away from the narrow expression range required for faithful repair, a problem that becomes especially difficult to interpret in neurons, where replication-linked strand cues are absent and mismatch repair can feed either correction or damage signaling depending on context. In a recent research paper published in a recent research paper published in Nucleic Acids Research and led by Professors John Tainer, Muralidhar Hegde from Houston Methodist Research Institute and The University of Texas MD Anderson Cancer Center, asked whether TDP43, long known for its central place in RNA handling and increasingly tied to amyotrophic lateral sclerosis and frontotemporal dementia, also controls this repair axis at the level of gene expression. That question follows naturally from two lines of biology that had not yet been brought fully into the same frame. One concerns TDP43 itself. The protein resides mainly in the nucleus, binds RNA and DNA, and helps govern splicing, transcript stability, transport, and autoregulation. The other concerns mismatch repair, whose core components MLH1, MSH2, MSH3, MSH6, PMS2, and PMS1 act through obligate heterodimeric assemblies to detect mismatches and initiate excision–resynthesis. The replication field has long treated mismatch repair as a major guardian of fidelity, but the same machinery can also participate in DNA damage signaling and, under certain expression states, contribute to mutagenic outcomes. That expression dependence matters because a repair pathway built from interacting subunits does not respond only to complete loss. Stoichiometric displacement can redirect pathway behavior as well.
The unresolved difficulty sat in the biology of nondividing cells. Cancer genetics had already established what mismatch repair deficiency does in proliferative tissues, but neuronal settings posed a different problem. In neurons, mismatch repair has been discussed in connection with deamination repair and repeat expansion, yet its operating logic has remained uncertain. The paper frames this uncertainty not as a marginal detail but as a central issue: if mismatch repair proteins must remain in balance, then any upstream regulator that controls their transcripts could alter genome handling in a way that standard DNA repair models would miss. That is where TDP43 became especially compelling. The group had already linked TDP43 proteinopathy to DNA double-strand breaks in neurons. Once one accepts that TDP43 governs large sectors of RNA metabolism, it becomes difficult to ignore the possibility that its impact on genome stability may run through repair gene transcripts themselves, not only through direct action at damaged chromatin. The motivation of the study came from exactly that gap. Rather than asking only whether TDP43 associates with DNA repair in a broad sense, the investigators asked a sharper mechanistic question: does TDP43 control mismatch repair gene expression, and if so, does it do this through the kinds of RNA-processing functions for which the protein is already known? That framing matters because it moves the problem away from a single lesion-repair event and toward regulatory architecture. If TDP43 governs splice choice and transcript persistence for selected mismatch repair genes, then TDP43 pathology could reshape repair capacity before any downstream DNA phenotype becomes visible. The study was built to test that regulatory idea across cultured cells, neuronal differentiation states, ALS-linked models, human disease tissue, and cancer datasets.
The investigators began with a broad DNA repair expression screen after reducing TDP43 in HEK293 cells, and mismatch repair stood out immediately. Lowering TDP43 to about half of control levels changed several repair-gene families, with MLH1, MSH3, MSH6, and PMS2 dropping by more than twofold at the transcript level; immunoblotting confirmed that the protein changes followed the same direction. They then moved in the opposite direction and raised wild-type TDP43 in differentiated SH-SY5Y cells. That reversal was informative because it established directionality: moderate TDP43 overexpression increased key mismatch repair factors above their basal levels. A regulator that shifts the pathway both downward and upward is doing more than marking cellular stress. It is actively setting the expression state of the pathway. They extended that logic into neural lineage models and used iPSC-derived neural progenitor stem cells, TDP43 knockdown again reduced MLH1, MSH3, MSH6, and PMS2. During differentiation from iPSC to neural progenitor to induced motor neuron, levels of MLH1, MSH2, and MSH3 fell in the motor-neuron state relative to progenitors, placing the TDP43–mismatch repair connection inside a cellular context where proliferative status changes. The paper makes clear that TDP43-dependent control of mismatch repair expression persists even when the cells are no longer defined by active cycling.
The group selected MLH1 and MSH6 for deeper analysis after in silico mapping identified transcript regions with strong predicted TDP43 binding. They designed exon–intron–exon assays so that product size would report intron retention directly. Once TDP43 was depleted, fully spliced MLH1 and MSH6 products dropped by more than half and unspliced variants increased. A minigene built from the MLH1 exon 17–intron 17–exon 18 region reproduced that effect, with efficient splicing in control cells and strong intron retention after TDP43 knockdown. They also examined a published Tdp43 CLIP-seq dataset to support direct transcript engagement. The design here was well chosen because it tied phenotype to transcript architecture: the reduction in mismatch repair proteins did not need to be inferred from global RNA disruption alone; it could be traced to specific processing defects in selected targets. The study then asked whether transcript decay contributes alongside splice disruption. Using α-amanitin in nondividing neuronal cultures, the investigators measured half-life changes after TDP43 depletion. MLH1 transcript half-life fell by 38%, and MSH6 fell by 56%, whereas TDP43 transcript half-life increased, fitting its known autoregulatory behavior. It means TDP43 stabilizes selected mismatch repair RNAs after processing. The regulatory effect, then, works through at least two layers: productive splicing and transcript persistence.
The in vivo and disease-linked experiments gave the mechanism biological reach. In one ALS-TDP43 mouse model that isolates nuclear localization sequence loss and cytoplasmic mislocalization, cortical Msh2 and Msh3 increased, with Msh6 also tending upward. In a second model with moderate CNS TDP43 overexpression, Mlh1 and Msh3 increased in males, and females showed increases in Mlh1, Msh2, and Msh3. Guamanian ALS brain tissue added the human disease correlate, with increased insoluble MLH1, MSH3, and MSH6. Functional tests connected those expression changes to DNA phenotypes: mismatch repair depletion prevented 6-thioguanine-mediated killing, TDP43 knockdown produced a similar protective effect, and in a TDP43NLS cell model, MSH2 knockdown partially reduced comet damage and γH2AX. The decision to target MSH2 was logically strong because MSH2 sits at the core of both MutSα and MutSβ complexes, so reducing it collapses pathway engagement broadly rather than trimming only one branch.
To sum up, the authors developed a mechanistic framework in which TDP43 regulates mismatch repair gene expression through selective control of transcript splicing and stability, with MLH1 and MSH6 defined as direct mechanistic examples. They combined expression profiling, exon–intron splice assays, a minigene reporter, transcript half-life measurements, ALS-linked mouse models, human ALS tissue analysis, and TCGA-based coexpression and mutational-burden analysis to track that regulatory axis across systems. What is technically distinct here is the integration of RNA-processing control with mismatch repair biology, rather than treating TDP43-associated genome instability only as a lesion-repair defect at the DNA level. We believe the scientific importance of the study comes from the way it changes where one looks for genome instability in TDP43 biology. A great deal of attention has centered on TDP43 aggregation, nuclear depletion, and repair defects at damaged DNA sites. This paper adds a different layer of control. It places mismatch repair gene regulation inside the TDP43 problem, with MLH1 and MSH6 emerging as especially clear transcript-level targets. Once that is recognized, genome instability linked to TDP43 no longer reads simply as a downstream consequence of proteinopathy. It also reads as a remodeling of repair-gene expression, driven through RNA processing and transcript maintenance.
That matters in neurodegeneration because mismatch repair in neurons has remained conceptually unsettled. The field has had reason to consider repair, repeat expansion, and damage signaling, yet it has lacked a persuasive upstream regulator that connects neuronal RNA dysregulation to mismatch repair stoichiometry. The present findings supply such a connection. They also make biological sense of why TDP43 pathology could produce DNA damage phenotypes that are not exhausted by double-strand break repair failure alone. When a regulator alters multiple mismatch repair components across dividing and nondividing cells, the downstream consequence need not be uniform loss of repair. It can be pathway miscalibration, with expression shifts changing how cells respond to mismatches, alkylation damage, and damage signaling inputs. The partial rescue seen after MSH2 depletion fits that logic closely: under TDP43 proteinopathy, mismatch repair activity can contribute to the DNA damage state rather than simply opposing it.
The cancer-facing implications are also substantial, though the paper keeps them grounded in association and coexpression analysis. Across TCGA tumor types, the authors found that TARDBP coexpresses strongly with selected mismatch repair genes, especially MSH6 and MSH2 in many cancers. They also report tumor classes in which higher TARDBP expression associates with greater mutational burden, and hierarchical clustering places MSH6 close to TARDBP and MSH2, with breast and lung tumors showing strong increases in mutational burden among patients overexpressing these genes. This is an interesting conceptual shift. The usual cancer discussion treats mismatch repair deficiency as the main genomic route to mutation load. Here, the paper makes room for a second expression-centered scenario in which elevated TDP43 and selected mismatch repair components track with heavier mutational burden. That does not erase the classical deficient-repair model. It adds a regulatory mode in which excess or misdirected mismatch repair activity may shape genome change in another way.
The broader methodological contribution is just as valuable. The study does not rely on one model or one readout. It moves from transcriptomics to immunoblotting, from splice assays to stability measurements, from cultured cells to mouse cortex and human CNS tissue, and then into cancer informatics. That breadth is not decorative. It is what allows the authors to argue that the TDP43–mismatch repair relationship is not a culture artifact confined to one transformed line or one neuronal preparation. The same regulatory theme appears across systems, and that consistency gives the mechanism weight. It also changes design logic for future studies of TDP43-associated disease. Repair pathways may need to be read not only through lesions and foci, but through the transcript-processing networks that set repair protein abundance in the first place.
A conservative reading of impact is still enough to show why this study will matter. It identifies TDP43 as an active regulator of mismatch repair gene expression, defines splicing and stability as operative mechanisms for at least two core transcripts, links that regulation to ALS-related pathology and human disease tissue, and extends the same axis into cancer-associated mutational patterns. That is a meaningful expansion of how genome maintenance can be thought about in TDP43-linked biology. It moves the conversation from damaged DNA alone to the RNA-level governance of the repair machinery that meets that damage.


Reference
Vincent E Provasek, Albino Bacolla, Suganya Rangaswamy, Manohar Kodavati, Joy Mitra, Issa O Yusuf, Vikas H Malojirao, Velmarini Vasquez, Gavin W Britz, Guo-Min Li, Zuoshang Xu, Sankar Mitra, Ralph M Garruto, John A Tainer, Muralidhar L Hegde, RNA/DNA-binding protein TDP43 regulates DNA mismatch repair genes with implications for genome stability, Nucleic Acids Research, Volume 53, Issue 18, 14 October 2025, gkaf920, https://doi.org/10.1093/nar/gkaf920
Go to Journal of Nucleic Acids Research